Biotechnology QMS [Quality Management System]


Quality in biotechnology is a critical aspect that determines whether products reach patients safely and consistently. Biotech organizations must prove that every activity—from research and development through manufacturing and distribution—occurs under control, with evidence that processes perform as intended and that products meet specifications every time. The stakes are high because most biotechnology products are derived from living systems and carry more inherent variability than traditional small molecules. A minor deviation can affect potency, purity, or safety, and regulators expect that organizations understand how each step influences the final product.
That level of control requires structure. That structure is the biotechnology Quality Management System (QMS).
The QMS provides a standardized framework that connects every operation to a common purpose: ensuring product quality and protecting patient safety.
It defines expectations for documentation, training, equipment, materials, data integrity, and decision-making. Without a QMS, even experienced teams struggle to maintain consistency when production scales, new technologies are introduced, or regulatory expectations shift. A well-defined system brings order to complex environments and keeps activities aligned with scientific, operational, and regulatory priorities.
An effective Biotechnology QMS allows organizations to demonstrate that they can manage the unique risks associated with biologics, cell and gene therapies, recombinant proteins, vaccines, and advanced modalities. These products demand heightened attention to contamination prevention, chain-of-identity verification, raw material traceability, cold chain integrity, and batch-to-batch consistency. A modern QMS provides the infrastructure needed to track and control these variables across the entire lifecycle.
What Is a Biotechnology Quality Management System (QMS)?
A biotechnology Quality Management System defines how an organization plans, executes, documents, monitors, and improves all activities that affect product quality, safety, and compliance. It integrates good manufacturing practices (GMP), good laboratory practices (GLP), and good clinical practices (GCP) into a single operational framework that supports regulatory expectations and scientific integrity.
The QMS is not a standalone function—it is the operational environment in which science, manufacturing, and quality oversight take place.
It connects research, production, laboratory operations, and supply chain into one structured ecosystem.
The biotech QMS serves as the operational foundation that supports compliance with FDA 21 CFR Parts 210 and 211, ICH Q7 for active pharmaceutical ingredients, ICH Q10 for pharmaceutical quality systems, and other GxP requirements. It ensures that materials, equipment, personnel, and data are managed under controlled, documented processes. Every batch, every record, and every decision must be traceable. Biotech products rely on precise biological interactions and often require complex manufacturing steps such as cell culture, fermentation, purification, and aseptic processing.
A QMS ensures these steps are controlled, monitored, and documented with the detail necessary to prove compliance.
A biotech QMS is not a single procedure or department. It is a connected network of policies, SOPs, records, training programs, change controls, deviations, CAPAs, supplier qualifications, and management reviews. Together, these elements create proof that operations meet quality standards and that risk is understood and managed throughout the product lifecycle. The QMS enables teams to communicate in a structured way so that scientific decisions, manufacturing activities, and investigations follow documented, controlled processes. Every action taken within a biotech organization leaves a traceable record within the QMS.
Modern biotech operations depend on digital traceability, real-time monitoring, and automated control. Manual systems, paper records, and isolated spreadsheets no longer support the complexity of biologics, cell and gene therapies, or large-scale fermentation. For this reason, biotech companies are shifting to validated electronic Quality Management Systems (eQMS) that combine regulatory compliance with operational intelligence. These digital systems reduce transcription errors, enforce controlled workflows, and automatically maintain audit trails. They bring clarity to complex operations where dozens of variables influence each step.
A modern eQMS centralizes quality processes, enforces data integrity, and provides visibility across development, manufacturing, and supply chain.
It eliminates fragmented tools, reduces human error, and helps organizations maintain GMP readiness at all times. With a digital QMS, teams access the same information from any location, investigate issues with complete context, and work from controlled documents that reflect the latest approved processes.
When deviations linger, when audits feel reactive, or when training and change control depend on email threads, the issue is not local—it is systemic. A biotech QMS provides the structure that keeps operations under control. It ensures decisions are documented, actions follow approved workflows, and investigations uncover the root cause rather than temporary fixes. The QMS becomes the backbone that supports scale, speed, and compliance.
Why a Biotech QMS Matters
Regulators evaluate biotechnology organizations not only by the quality of their products but by the maturity of their systems. A QMS demonstrates that the organization can consistently produce safe and effective products under controlled conditions. It turns regulatory requirements into day-to-day practice. Without a strong QMS, even a well-designed process becomes vulnerable to deviations, data integrity issues, and inconsistent outcomes.
The importance of a QMS in biotechnology extends far beyond regulatory fulfillment.
It reduces waste, prevents contamination, improves batch success rates, and protects patient safety. It aligns cross-functional teams that often operate with different scientific or operational priorities. It ensures that knowledge gained during development is preserved through scale-up and commercialization. And it keeps organizations prepared for inspections at any time.

An effective QMS matters in five key ways.
1. Compliance with GMP and GxP Requirements
The FDA, EMA, and other global regulators expect that biotech manufacturers maintain a documented, controlled quality system. FDA 21 CFR Parts 210 and 211 define current Good Manufacturing Practice (cGMP) for drugs and biologics. ICH Q10 provides the framework for a pharmaceutical quality system that promotes continual improvement. Together, they require that organizations:
- Define and follow written procedures
- Maintain accurate, controlled records
- Validate critical processes
- Qualify and train personnel
- Control suppliers and materials
- Investigate deviations and implement CAPA
- Conduct management review and ensure oversight
A QMS operationalizes these expectations, ensuring that regulatory principles become measurable, repeatable actions. Inspections focus on how well the QMS functions in real time, not simply whether documents exist. Inspectors may request to see deviation histories, training effectiveness evaluations, CAPA closure times, and audit trails from equipment logs or batch records. They want to confirm that quality is integrated into operations and not managed reactively.
Compliance is not just about avoiding observations. It supports investor confidence, strengthens partnerships, and enables market expansion.
When organizations demonstrate strong QMS maturity, they reduce the time and cost associated with regulatory submissions because data is already structured, traceable, and complete.
2. Risk Reduction
Risk in biotechnology extends beyond process failure—it includes potential harm to patients, contamination of product, or loss of data integrity. Without a system, the likelihood of uncontrolled changes, incomplete documentation, and unqualified equipment increases. Even minor issues such as a temperature excursion, mislabeled sample, or incomplete environmental monitoring form can escalate into large-scale investigations.
A QMS integrates risk management across the lifecycle, using ICH Q9 and ICH Q10 principles to identify, evaluate, and mitigate risks in development, production, and distribution.
It ensures every deviation, change, and supplier issue is assessed for potential quality impact. The resulting documentation becomes both internal justification and regulatory evidence.
Risk management becomes especially important for advanced therapies. Cell and gene therapies require chain-of-identity protection. Biologics demand control of upstream and downstream parameters that influence expression, yield, and impurity profiles. Viral vector production requires contamination prevention and segregation. A QMS connects all these risks and ensures that mitigation strategies are documented, monitored, and verified.
Reducing risk improves batch success rates and reduces costs associated with lost materials, failed lots, or extended investigations. It also strengthens confidence in data used for regulatory filings, clinical trials, or product release.
3. Efficiency and Time to Market
Quality does not delay progress; poor systems do. Manual reviews, disconnected spreadsheets, and redundant approvals slow production and increase the risk of rework. In biotechnology, delays can disrupt clinical timelines, affect supply continuity, and increase the cost of goods.
An eQMS streamlines communication and ensures teams work from the same controlled record.
Design transfers, batch reviews, and deviations close faster because the information is centralized and traceable. Real-time access to records allows faster decision-making and supports earlier release of compliant batches, improving speed without compromising quality.
Efficiency also improves cross-functional collaboration. R&D teams can transfer knowledge to manufacturing with clarity because information is structured and stored within the QMS. Operations teams can review process histories without searching through legacy systems. QA can approve changes, review investigations, and evaluate risk faster because workflows enforce stepwise, documented actions.
The QMS becomes a driver of operational agility. Faster response times, improved documentation accuracy, and fewer process interruptions all contribute to shorter timelines and more reliable production.
4. Traceability and Data Integrity
Every step in biotech manufacturing must be traceable. From cell line development to final lot release, each decision must be documented and attributed. Inspectors expect complete data histories showing how raw materials, in-process controls, and final results link together.
A biotech QMS ensures that traceability is built into every process. Audit trails, version control, and electronic signatures create the documented chain of evidence that proves compliance.
Data integrity principles—ALCOA+ (Attributable, Legible, Contemporaneous, Original, Accurate, Complete, Consistent, Enduring, and Available)—are enforced through system design.
Traceability is essential in biologics where variability is inherent. Raw material lots can influence yield. Cell line performance can drift. Environmental conditions can affect potency. The QMS helps track these factors so that investigations have complete context and so that trends can be identified early.
Strong data integrity also protects the organization from compliance risk. Regulators have increased scrutiny on electronic data, incomplete records, late entries, and uncontrolled spreadsheets. A QMS enforces access control, time-stamped audit trails, and versioned document histories to ensure that data remains trustworthy throughout its lifecycle.
5. Continuous Improvement
Continuous improvement is not a slogan; it is a regulatory expectation under ICH Q10. A mature QMS transforms raw data into actionable insight, helping leadership identify recurring deviations, training gaps, or supplier issues before they escalate. Improvement efforts become systematic rather than reactive.
The QMS centralizes information so that trends can be monitored in real time.
Deviations can be categorized and analyzed. CAPA effectiveness can be evaluated through defined verification steps. Supplier performance can be assessed using quality metrics. Training gaps can be identified by comparing required qualifications with SOP changes or audit findings.
Continuous improvement strengthens operational readiness. It reduces the number of batch failures, improves resource planning, and helps organizations respond to regulatory changes quickly. When teams use data to guide improvement rather than waiting for issues to surface, compliance becomes a predictable outcome.
Biotech QMS Standards and Regulatory Framework
A biotech QMS operates under global regulatory frameworks designed to ensure product quality, patient safety, and data integrity. These frameworks define the principles and expectations that organizations must meet to manufacture, test, store, and distribute products consistently. Understanding these regulations is essential because they shape how procedures, controls, and documentation are structured.
1. FDA 21 CFR Parts 210 and 211
Parts 210 and 211 outline current Good Manufacturing Practice (cGMP) for drugs and biologics in the United States. They cover personnel qualifications, facilities, equipment, production, records, and laboratory controls. These regulations establish minimum standards that all manufacturers must follow to ensure product quality.
Biotech organizations must establish procedures for every activity that affects product quality. Records must demonstrate that each batch is produced and controlled in accordance with written instructions.
Batch production records, deviations, and laboratory data must all be reviewed and approved before release. These requirements ensure batch-to-batch consistency and provide traceability.
The regulations give inspectors authority to examine any part of the QMS, including training files, equipment logs, environmental monitoring data, and deviation histories. A QMS aligns operations with these expectations by standardizing how data is captured, reviewed, and approved.
2. ICH Q7: GMP for Active Pharmaceutical Ingredients
ICH Q7 applies to biotech companies that manufacture APIs, including those produced by fermentation or cell culture. It defines expectations for equipment design, cleaning, contamination control, validation, and quality oversight.
For biotechnology, this includes controls on cell banks, viral clearance, and contamination prevention in upstream and downstream processing. A QMS ensures that these processes are validated, monitored, and documented. ICH Q7 requires organizations to establish controls that reduce contamination risk and ensure that product quality is maintained through each step of production.
3. ICH Q10: Pharmaceutical Quality System
ICH Q10 provides the model for a modern pharmaceutical QMS. It integrates quality management throughout the product lifecycle, emphasizing management responsibility, continual improvement, and risk-based decision-making.
Under ICH Q10, a biotech QMS must enable process performance monitoring, CAPA management, and knowledge management.
The goal is not only compliance but sustainable quality throughout commercial operations. ICH Q10 encourages scalability and harmonization so that organizations can adapt as they grow or shift operations between sites.
4. GxP Integration
Biotech companies often operate under multiple “GxP” standards—GMP for manufacturing, GLP for laboratories, and GCP for clinical studies. A unified QMS integrates these systems so that information, materials, and responsibilities remain traceable across stages.
For example, analytical data produced under GLP may support GMP decisions during manufacturing. Clinical results governed by GCP may influence process controls or batch release strategies. A QMS connects these domains so that transitions between phases remain compliant and traceable.
5. Data Integrity Guidance
Data integrity is central to every regulation. FDA, EMA, and MHRA guidance all require that electronic data be attributable, accurate, complete, and accessible throughout its retention period.
An eQMS enforces these principles through audit trails, secure access, and version control.
It ensures that data generated in manufacturing or testing remains reliable and that records cannot be altered without traceability. Regulators expect that systems controlling data have defined user roles, time-stamped records, automated audit logs, and protection against unauthorized modification.
Documentation Structure of a Biotech QMS
Documentation is the visible proof of control. Inspectors evaluate both content and structure to confirm that the organization’s processes are defined and followed. A QMS without documentation is not a functioning system. Every activity must be traceable through documented evidence.
1. Quality Manual
The Quality Manual defines the scope of the QMS and maps processes to regulatory requirements. It explains how the organization meets cGMP, ICH Q10, and local health authority expectations. It sets the tone for the organization’s quality culture and aligns teams on the principles that guide daily operations.
The Quality Manual also provides a high-level overview of key processes, governance structures, and the interactions between departments. It is often one of the first documents requested during inspections because it provides context for how the rest of the system functions.
2. Policies
Policies express management’s commitment to quality, data integrity, and patient safety. They define expectations for risk management, supplier oversight, and change control. Policies are broad, stable documents that reflect the organization’s values and regulatory obligations.
Policies serve as the foundation for procedures and ensure that quality decisions align with corporate priorities. They clarify responsibilities and establish boundaries for acceptable practices. Policies also guide decisions when procedures are unclear or not yet updated.
3. Standard Operating Procedures (SOPs)
SOPs describe how regulated activities are performed. In biotechnology, SOPs typically cover:
- Equipment cleaning and sterilization
- Environmental monitoring and contamination control
- Material management and batch manufacturing
- Deviation, CAPA, and change control
- Validation and calibration
- Document control and training
- Batch record review and release
Each SOP must be controlled, versioned, and approved by authorized personnel. SOPs ensure that processes are performed consistently regardless of who completes the task. They reduce variation and provide the foundation for training and qualification.
4. Work Instructions and Forms
Work instructions provide detailed steps for specific tasks, such as performing an aseptic fill, running a chromatography column, or verifying instrument calibration. They offer clarity on how procedures should be executed at the task level.
Forms standardize how information is recorded, ensuring that critical data are captured consistently and legibly. They reduce variation in documentation and help ensure that critical fields are not missed during data entry.
Work instructions and forms ensure that procedures translate into accurate execution. They also simplify investigations by providing clear evidence of what occurred during each process step.
5. Records
Records prove that procedures were followed. They include:
- Batch production records
- Equipment logs
- Training files
- Deviation and CAPA reports
- Stability studies and validation summaries
- Supplier qualification documents
Records must be complete, contemporaneous, and available for the defined retention period. Electronic records must comply with 21 CFR Part 11 and EU Annex 11. These records form the basis of regulatory inspections and must reflect the actual activities performed by personnel.
Core Processes in a Biotech QMS
A QMS is only effective when it controls the processes that influence product quality. Each process must be defined, implemented, and periodically evaluated for performance. These processes work together to form a cohesive system that governs all quality-related activities.
1. Document Control
Document control governs creation, approval, distribution, and revision of all controlled documents. It ensures that only current versions are in use and that obsolete documents are withdrawn.
Electronic document control systems support version tracking, approval routing, and audit trails. Inspectors often begin with document control during audits because it reflects the health of the entire system. Ineffective document control can lead to inconsistent practices, outdated procedures, and compliance risk.
2. Change Management
Change management ensures that modifications to equipment, materials, or processes do not introduce risk. Each proposed change is documented, risk assessed, reviewed, and approved before implementation.
In biotech manufacturing, even small changes—such as adjusting media formulation or altering cleaning parameters—can affect product quality. A formal process ensures evaluation and traceability. Change control also supports product lifecycle management by ensuring that knowledge gained during development is incorporated into commercial operations in a controlled manner.
3. Training Management
Personnel must be qualified before performing regulated tasks. Training management links job roles to required SOPs and tracks completion.
The QMS must ensure that employees are trained on current versions of procedures and that training effectiveness is verified. Training records often serve as the first evidence requested by inspectors. Proper training prevents human error and ensures that procedures are executed consistently.
4. Risk Management
Risk management applies across all operations, from development to production. It involves identifying hazards, evaluating probability and impact, implementing controls, and monitoring residual risk.
ICH Q9 provides a framework for structured risk assessment. Integration with CAPA and change management ensures that new information continually updates the risk profile. Risk management strengthens decision-making, reduces uncertainty, and helps prioritize improvement efforts.
5. Deviation and CAPA Management
Deviations capture any departure from approved instructions or expected results. CAPA ensures that root causes are identified and eliminated.
An effective CAPA process includes:
- Problem definition
- Investigation and root cause analysis
- Implementation of corrective actions
- Verification of effectiveness
CAPA records provide a clear trail from detection to resolution and are a primary focus during regulatory audits. CAPA effectiveness checks ensure that actions prevent recurrence and that improvements are sustained.
6. Supplier and Material Management
Biotech production relies on raw materials, consumables, and services provided by external partners. Supplier management ensures that these inputs meet predefined quality standards.
Processes include supplier qualification, performance monitoring, and periodic re-evaluation. Supplier agreements must clearly define responsibilities for testing, release, and change notification. Poor supplier performance can affect product quality, increase variability, and cause batch failures.
7. Production and Process Control
Production processes must operate under validated and controlled conditions. Equipment calibration, cleaning, and maintenance must be documented.
The QMS ensures that batch records reflect actual production data and that deviations are documented in real time. In-process controls verify that parameters remain within defined limits. Production control is essential in biotechnology where upstream and downstream processes involve sensitive systems.
8. Validation
Validation provides documented evidence that processes, methods, and systems perform as intended. It includes equipment qualification (IQ/OQ/PQ), cleaning validation, process validation, and computer system validation.
Each validation activity must follow an approved protocol and produce a report that confirms acceptance criteria were met. A validated state ensures consistency and predictability.
9. Internal Audits and Management Review
Internal audits test whether the QMS is effective and compliant. Audit results feed into CAPA and management review.
Management review consolidates metrics, audit findings, and improvement initiatives. It allows leadership to assess QMS performance and allocate resources where needed. Internal audits ensure that the system operates as intended and that gaps are identified early.
The Biotech QMS Across the Product Lifecycle
A biotech QMS extends beyond manufacturing. It connects every stage from development through post-market monitoring. This holistic approach ensures that quality principles apply throughout the lifecycle.
1. Research and Development
In early R&D, the QMS governs data recording, experiment reproducibility, and material traceability. Documentation ensures that research results support later regulatory submissions.
Quality oversight in R&D may include controlled notebooks, data integrity checks, and versioned protocols. These controls ensure that development decisions are traceable and justifiable.
2. Technology Transfer
When moving from development to manufacturing, the QMS ensures knowledge transfer, defines critical process parameters, and verifies process readiness. Technology transfer packages document experiments, process parameters, and controls.
The QMS ensures that development knowledge is structured, reviewed, and approved so that manufacturing can replicate processes reliably.
3. Manufacturing and Scale-Up
During scale-up, the QMS monitors process capability and enforces change control. It ensures that pilot-scale learnings transfer to commercial scale without deviation from validated parameters.
Scale-up activities require extensive documentation, process characterization, and control strategy development. The QMS manages these activities to maintain consistency.
4. Distribution and Storage
The QMS defines how products are packaged, labeled, and stored to maintain stability. Transportation conditions must be monitored, and deviations must trigger investigation.
Chain-of-custody and cold chain processes are critical for many biotech products. The QMS ensures compliance with storage and distribution requirements.
5. Post-Market Monitoring
After release, post-market data such as complaints, stability results, and adverse event reports feed back into risk management and process improvement. This feedback loop supports continual compliance with GMP and ICH Q10.
Post-market surveillance helps identify potential product issues early and ensures that corrective actions are implemented.
Roles and Responsibilities in the Biotech QMS
A QMS functions effectively only when roles and responsibilities are defined, documented, and understood across the organization. Everyone contributes to quality.
Executive Management sets policy, provides resources, and ensures the system operates as intended. They review metrics and trends to verify that processes remain effective. Management accountability is a regulatory expectation under ICH Q10 and 21 CFR Parts 210 and 211.
Quality Assurance (QA) owns oversight of controlled processes. QA reviews and approves deviations, batch records, and validation reports. It coordinates audits, manages CAPA, and ensures training and documentation meet regulatory expectations.
Quality Control (QC) manages laboratory testing, data review, and method validation. QC ensures that test results are accurate, complete, and traceable. It verifies that materials and finished goods meet specifications before release.
Regulatory Affairs ensures compliance with regional and global regulations. This function manages submissions, monitors changes in regulatory requirements, and maintains licenses and certificates.
Manufacturing and Operations execute production activities according to validated procedures. They document all steps in real time and report deviations immediately.
Research and Development ensures that design and process knowledge transfer seamlessly to production. They maintain design control and validation documentation aligned with GMP requirements.
Supply Chain and Procurement qualify suppliers, maintain approved vendor lists, and ensure that materials meet defined acceptance criteria.
IT and Systems Owners maintain validated systems that support the QMS. They control access, manage backups, and ensure compliance with 21 CFR Part 11 and EU Annex 11.
Each employee is responsible for following current procedures, reporting quality issues, and maintaining awareness of their training status. When accountability is clear, compliance becomes measurable.
Implementing a Biotech QMS
Building or modernizing a QMS requires structure. A defined plan ensures that the system supports both regulatory expectations and business goals.
1. Define Scope and Objectives
Determine which activities the QMS will cover, such as clinical manufacturing, commercial production, or contract testing. Identify applicable regulations—FDA cGMP, EMA guidelines, ICH Q7 and Q10, and any regional health authority requirements.
Defining scope ensures that the QMS covers all necessary processes without introducing unnecessary complexity.
2. Conduct a Gap Assessment
Compare current practices against regulatory and ISO expectations. Document where procedures, records, or controls are missing. Prioritize gaps based on risk and business impact.
The gap assessment provides a roadmap for improvement and clarifies which processes need immediate attention.
3. Develop the Documentation Framework
Define the hierarchy of documents: policies, SOPs, work instructions, and records. Establish document templates and naming conventions for consistency.
A well-structured documentation system reduces confusion and improves efficiency.
4. Select Technology
Choose a QMS platform that supports your operational model. For biotech, electronic systems must handle both regulated manufacturing and development data, maintain audit trails, and allow secure role-based access.
The right technology minimizes manual work and improves traceability.
5. Validate the System
System validation demonstrates that the eQMS performs as intended. This includes user requirements, testing, and documented evidence of compliance with Part 11 and Annex 11.
Validation ensures that electronic systems are trustworthy and compliant.
6. Train and Deploy
Training ensures that users understand how to operate within the QMS. Deployment should follow a phased approach, starting with core processes such as document control, CAPA, and training before expanding to supplier and audit management.
Training and phased deployment reduce errors during transition.
7. Monitor and Adjust
Once operational, the QMS must be reviewed periodically. Metrics, internal audits, and management reviews identify improvement opportunities and confirm ongoing compliance.
A QMS must evolve as operations grow and regulatory expectations change.
Common Challenges in Biotech QMS Implementation
Several recurring challenges affect biotech companies as they build or scale quality systems. Addressing these early prevents long-term inefficiency.
1. Fragmented Processes
Departments often maintain independent systems or local practices. Without integration, data becomes inconsistent and traceability breaks down. Fragmented processes increase the risk of errors and complicate investigations.
2. Manual or Hybrid Systems
Paper records and spreadsheets introduce errors and limit visibility. Manual approvals slow change control and batch release. Hybrid systems make it difficult to maintain data integrity because workflows depend on human intervention.
3. Inconsistent Training
Training systems that are not linked to document control cause personnel to work from outdated procedures. Inconsistent training increases the risk of errors and slows investigations.
4. Uncontrolled Change
Process or equipment changes implemented without risk assessment lead to compliance gaps. Uncontrolled changes can affect product quality and trigger regulatory observations.
5. Reactive CAPA
Organizations that treat CAPA only as a regulatory requirement fail to use it as a tool for improvement. Reactive CAPA leads to repeated issues and longer investigation times.
QMS Metrics and Performance Monitoring
A functioning QMS must generate measurable results. Metrics confirm whether the system performs as intended and guide management review.
Metrics help evaluate whether processes are effective, compliant, and aligned with regulatory expectations. They also highlight areas that need improvement.
Typical metrics include:
- Number and closure time of deviations and CAPA
- Rate of recurring nonconformances
- Training completion rates
- Audit findings by severity
- On-time supplier requalification
- Batch rejection rates
- Complaint rates normalized to units released
Metrics provide clarity and ensure that leadership can evaluate the health of the QMS. They support strategic decision-making by highlighting inefficiencies and emerging risks.
Integration of the QMS with Other Systems
A biotech QMS rarely operates alone. It must exchange data with systems that control materials, design, and laboratory results. Integrated systems reduce duplication, improve accuracy, and strengthen traceability.
- Enterprise Resource Planning (ERP) connects material control and production planning with quality data.
- Laboratory Information Management Systems (LIMS) capture analytical results and feed them into batch disposition and stability programs.
- Manufacturing Execution Systems (MES) document in-process activities, equipment usage, and environmental conditions.
- Customer Relationship Management (CRM) systems support complaint handling and post-market surveillance.
Integration reduces manual transcription, supports real-time insight, and streamlines batch release.
Continuous Improvement and Management Review
Continuous improvement ensures that the QMS adapts to changing conditions and new knowledge.
Management review consolidates data from audits, CAPA, deviations, complaints, and process metrics. The leadership team evaluates performance, assesses effectiveness, and approves improvement actions.
Continuous improvement strengthens operational discipline and prepares organizations for regulatory changes.
Maintaining the Biotech QMS Over Time
Regulations and technologies change. The QMS must evolve to remain compliant and relevant.
Periodic reviews ensure that procedures match current operations. New regulations such as updates to ICH Q9 or guidance on data integrity must trigger impact assessments and updates to relevant documents.
Personnel changes require updated role descriptions and training. Mergers or new facilities demand harmonized processes and document control.
Sustaining the QMS requires ongoing commitment. It is not a one-time project but a continuous discipline supported by data and leadership oversight.
Digital Transformation and the Modern Biotech QMS
As biotech products grow in complexity, digital systems are now essential to maintain control and agility.
1. Electronic QMS (eQMS)
An eQMS replaces paper with a validated, digital environment for managing documents, training, CAPA, and change control. It automates workflows, maintains audit trails, and ensures compliance with Part 11.
2. Data Connectivity
A modern eQMS connects with ERP, LIMS, and MES systems, creating a unified data model across the product lifecycle. This linkage provides real-time visibility of process performance, quality trends, and risk indicators.
3. Predictive Analytics
Quality data from multiple processes can be analyzed to identify early warning signs of deviation. Predictive analytics supports proactive decision-making and reduces batch failures.
4. AI and Automation
AI supports document search, trend recognition, and process optimization within validated boundaries. Automation streamlines repetitive tasks such as training assignment, approval routing, and audit scheduling.
5. Compliance and Security
Digital systems must maintain data integrity. Access controls, encryption, and audit trails protect information and ensure traceability.
Validated systems help organizations maintain inspection readiness at all times by ensuring that information is complete and retrievable.
The Future of Biotech Quality Management
The future of biotech quality management is shaped by automation, analytics, and a shift toward continuous verification.
Processes will rely less on manual review and more on real-time monitoring. Sensors, integrated systems, and AI will track process parameters continuously, triggering alerts before deviations occur.
Knowledge management will become part of daily operations, with data flowing seamlessly between research, development, and manufacturing.
Regulators are already signaling support for innovation through initiatives like FDA’s Emerging Technology Program and ICH Q12 on lifecycle management. These frameworks encourage modernization while maintaining control.
Organizations that build flexible, data-driven QMS frameworks today will adapt more easily as technologies and regulations advance.
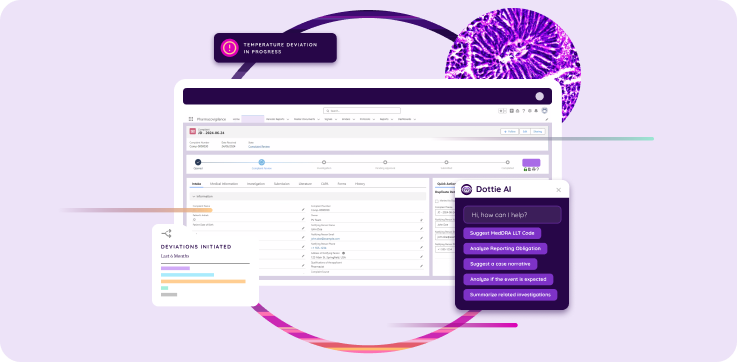
How Dot Compliance Supports Biotech Quality
Dot Compliance provides an electronic Quality Management System designed for biotech and pharmaceutical organizations that operate under GMP and GxP regulations.
Built on Salesforce, the platform delivers a validated, secure environment that unifies key quality processes, including:
- Document control and training management
- Deviation, nonconformance, and CAPA
- Supplier and audit management
- Risk and change control
- Complaint handling and post-market monitoring
- Batch release and review workflows
The system maintains full compliance with 21 CFR Parts 210–211, Part 11, EU Annex 11, and ICH Q10.
With integrated AI through Dottie AI, users can search records, summarize data, and identify trends directly within the controlled environment. Every query and output remains auditable and secure.
Dot Compliance supports biotech organizations in achieving inspection readiness, improving efficiency, and scaling operations globally without compromising data integrity.
Frequently Asked Questions (FAQs): Biotechnology Quality Management Systems
What is a Biotechnology Quality Management System (QMS)?
A biotech QMS is the structured framework that governs how an organization plans, executes, documents, monitors, and improves all activities that affect product quality, patient safety, and regulatory compliance. It spans research, development, manufacturing, distribution, and post-market monitoring, ensuring that all work follows controlled processes and that records provide complete traceability.
Why is a QMS important in biotechnology?
Biotech products are complex, variable, and sensitive to minor changes in process or environment. A QMS provides the structure needed to control this complexity. It reduces risk, supports compliance with global regulations, improves batch success rates, strengthens documentation, and keeps organizations inspection-ready at all times.
Which regulations apply to a biotech QMS?
A biotech QMS must satisfy FDA 21 CFR Parts 210 and 211, ICH Q7, ICH Q10, and applicable GxP requirements across manufacturing, laboratory operations, and clinical work. It must also comply with data integrity expectations and electronic records standards such as 21 CFR Part 11 and EU Annex 11.
What documentation is required in a biotech QMS?
A complete QMS includes a Quality Manual, policies, SOPs, work instructions, controlled forms, and operational records. These documents establish how work is performed and provide objective evidence that procedures were followed, data are accurate, and decisions were justified.
What are the core processes of a biotech QMS?
Essential processes include document control, training, change management, risk management, deviation and CAPA handling, supplier qualification, production and process control, laboratory oversight, internal audits, and management review. Together, these processes maintain stability, traceability, and compliance across the lifecycle.
How does a QMS support the biotech product lifecycle?
A QMS connects early research, tech transfer, scale-up, commercial manufacturing, distribution, and post-market monitoring. It ensures that knowledge is preserved, changes are controlled, risks are managed, and quality data remain traceable across each stage.
What challenges do biotech companies face when implementing a QMS?
Common challenges include fragmented processes, reliance on manual or hybrid systems, inconsistent training, uncontrolled changes, and reactive CAPA. These issues lead to data integrity gaps, longer investigations, and increased inspection risk.
How do organizations measure whether their QMS is performing well?
Performance is tracked through metrics such as deviation and CAPA cycle time, recurring issues, training completion rates, audit findings, supplier performance, batch rejection rates, and complaint trends. These metrics help identify risks and guide improvement.
Should a biotech QMS integrate with other systems?
Yes. Integration with ERP, LIMS, MES, CRM, and other enterprise systems reduces manual work, strengthens traceability, and improves batch release efficiency. Integrated environments provide a complete view of quality data across operations.
Why are electronic QMS (eQMS) platforms important for biotech?
Digital systems enforce data integrity, automate workflows, maintain audit trails, and centralize critical processes. They reduce transcription errors, improve visibility, and support compliance with 21 CFR Part 11 and Annex 11—capabilities that manual systems cannot provide.
How is AI being used in modern biotech QMS environments?
AI supports document retrieval, trend analysis, and issue investigation within validated, governed frameworks. It accelerates quality workflows by improving access to information while preserving traceability and compliance.
How does Dot Compliance help biotech organizations strengthen their QMS?
Dot Compliance provides a cloud-based, Salesforce-native eQMS that unifies document control, training, deviations, CAPA, supplier management, complaints, audits, and quality events. The platform supports Part 11 and Annex 11 compliance, integrates with ERP/LIMS/MES systems, and includes governed AI through Dottie AI to help teams work more efficiently while maintaining full traceability.
Conclusion
Quality is not a department. It is a system that connects every function, every record, and every decision. In biotechnology, where products affect patient safety and often involve complex biological systems, a strong QMS is the foundation of trust.
Manual processes cannot support today’s regulatory and operational demands.
A modern, validated eQMS enables traceability, compliance, and continuous improvement at scale.
Organizations that invest in a connected, data-driven QMS position themselves for sustainable growth, faster approvals, and long-term compliance.
Dot Compliance provides the tools and structure to make that possible.
Book a demo to see how a modern eQMS can strengthen your biotech operations and keep your quality system inspection-ready every day.