Quality Management System (QMS) for Medical Device


Maintaining compliance with safety and quality standards is a significant issue in medical device manufacturing. Millions of dollars are lost by manufacturers every year due to regulatory non-compliance issues and related lawsuits. We continuously hear of infractions and warnings issued to companies for lack of regulatory compliance. Moreover, the increased incidences of diagnostic errors have given rise to further regulation and standardization of diagnostic equipment, as well as the ongoing evolution of regulations.
Patients and medical professionals have justifiably high expectations for their devices’ safety, quality, and efficacy. Government regulators have been doing their best to ensure that patients receive the safest and most efficient device possible.
The two most relevant quality regulations for medical device manufacturers are the FDA 21 CFR Part 820 and the ISO 13485:2016. Complying with these requires constant vigilance and automated solutions that keep up with the ongoing evolution of the regulatory market. That’s why manufacturers deploy Quality Management Systems to enable standardization and certification to meet regulatory standards.
In this post, we will take a deep dive into the medical device quality management system, look at the principal regulations governing the QMS, the difference between a QMS and an eQMS, and how a robust eQMS can solve the regulatory quagmire for medical device manufacturers.
What is a medical device QMS?
QMS, or quality management control, supports device manufacturers in improving and maintaining the quality of their medical devices and related services, as well as constantly and consistently meeting customer and regulatory requirements.
A medical QMS is a structured system that documents the procedures and processes throughout the lifecycle of a medical device:
- Design
- Process and production control, including equipment product surveillance, including risk monitoring
- Vigilance and complaints
- Resources including training and work environment
- Management responsibility, including readiness, review, and audit
- Corrective and preventive action
- Change management, including risk review
The QMS supports the manufacturers, leading to ISO confirmation and FDA compliance.
Though often used interchangeably, it is essential to differentiate the ”traditional” QMS from the eQMS, or electronic Quality Management System, which uses advanced IT infrastructures and technologies to provide the same functions in a more efficient process and avoid unnecessary mistakes. The eQMS is a digital approach to QMS–managing and documenting the business processes for quality, compliance, and product development across the entire life sciences value chain.
What are the main regulations governing medical device QMS?
In the medical device manufacturing business, safety and quality are non-negotiable. Accordingly, the International Organization for Standardization (ISO) and the American Food and Drug Administration (FDA) developed their own regulations governing quality management for medical devices.
The ISO developed ISO 13485, a stand-alone quality management standard (QMS) specific to the medical device industry. The FDA 21 CFR Part 820 is the mandatory standard for medical devices distributed in the US.
The ISO 13485: 2016 standard sets standards for quality management systems in which a company, regardless of size or type, must demonstrate its capacity to consistently offer medical devices and related services that fulfill customer and regulatory requirements. Within the EU, the manufacturer can voluntarily choose to conform to ISO 13485:2016 or can be required to conform by contract.
The FDA 21 CFR Part 820 is similar to the ISO 13485:2016. The main differences between the two are :
- The ISO is the international standard, while the FDA is mandatory for the US
- The ISO prioritizes a risk-based approach, while the FDA rarely specifies risk management requirements
- The ISO 13585 has undergone multiple revisions with various countries collaborating with other ISO members, while the FDA 21 CFR Part 820 was created solely by the FDA. It has retained the same structure since 1997 with various additions throughout and was created solely by the FDA.
Most interestingly, on February 23rd, 2022, the FDA published a proposed regulation, to amend the medical “device current good manufacturing practice requirements of the Quality System (QS) regulation (21 CFR Part 820) to incorporate the international standard specific for medical device quality management systems set by the International Organization for Standardization (ISO), ISO 13485:2016 Medical devices – Quality management systems – Requirements for regulatory purposes.”
In other words, the FDA is working toward aligning its regulatory framework with the ISO. This endeavor aims to promote consistency in the regulation of devices and the timely introduction of safe, effective, high-quality devices for patients.
A Step-by-step Guide to Medical Device Quality System Management
Now, let’s break down the steps of a medical device QMS.
First, we will look at this through the ISO 13485:2016, and then via FDA 21 CFR Part 820. Keep in mind that while both have the same goals, they are differently worded documents from different parts of the world.
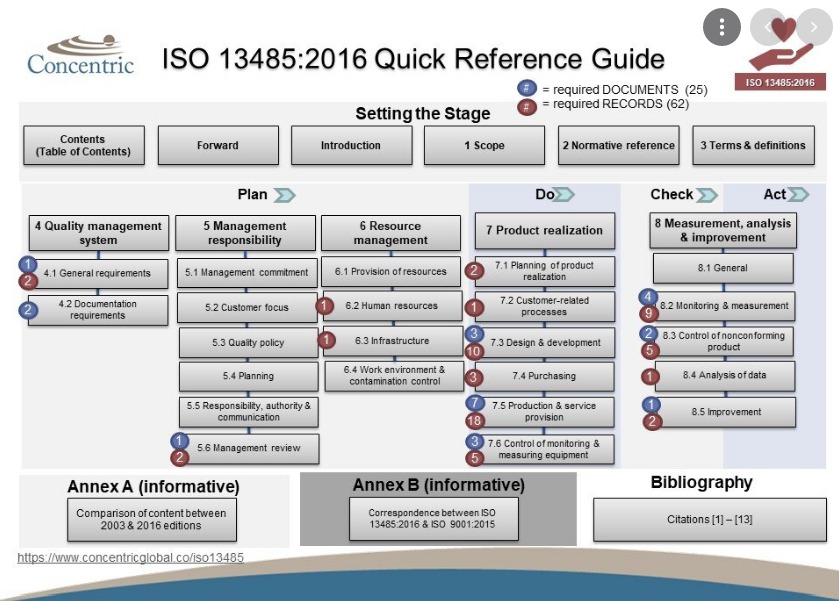
QMS According to ISO 13485:2016
Documentation Requirements
These requirements include:
- Quality Manual
- Medical Device File
- Document Control
- Control of Records
The Quality Manual describes the entire scope of the QMS, including any clauses that are excluded or non-application supported with justification, lists the procedures comprising the QMS, describes the interaction of the QMS, and outlines the structure of QMS documentation.
The Medical Device File must contain the following information:
- The product’s description, including its intended usage and warnings
- Instructions for use and product labeling
- Product features and specifications
- Manufacturing, inspection, labeling, packing, storage, handling, and distribution specifications and processes
- Measurement and monitoring specifications
- Product installation specifications and processes (if applicable)
- Product-servicing procedures (if applicable)

A Document Control procedure defines your company’s criteria for document control, including ensuring documents are reviewed and approved before implementation, means to revise documents and identify changes, and that current versions are available at the point of use.
Control of Records is evidence that certain processes have been followed. When it comes to review and approval, the same requirements apply.
Management Responsibility
Top management must be involved in the implementation and maintenance of the QMS. Several stages go into the standardization of management oversight. They are:
Quality Policy
Top management must make a commitment to comply with requirements and maintain the effectiveness of the quality management system by creating a quality policy.
Quality Management System Planning
Top management is responsible for the quality management system to meet all requirements and quality objectives.
Responsibility, Authority, and Communication
The ultimate responsibility to communicate with the authorities falls on the company’s management team. This includes ensuring that processes needed for the quality management system are documented and that the necessary procedures are communicated within the company.
Management Review
The organization is required to document procedures for management review. To verify its suitability, sufficiency, and effectiveness, top management examines its quality management system at recorded planned intervals. The assessment of potential for improvement and the need for modifications to the quality management system, including the quality policy and quality targets, will be included in the evaluation.
Resource Management
This step covers all the necessary requirements for the resources used to manufacture the device. This includes the people involved in the process (emphasizing training and competency, documenting training plans, and maintaining objective evidence of training).
It also includes infrastructure, processes, overall work environment, as well as buildings, workspaces, process equipment, software to support business operations, and support services. The infrastructure must be appropriate for the types of activities and operations conducted.
Product Realization Planning
This step discusses how a company develops, manufactures, and distributes medical devices. It covers the resources and processes needed to define client needs, design and development, purchasing, manufacturing, and field service.
It is essential to ensure that quality criteria are established for the products and that required processes and supporting documentation are defined. Requirements for infrastructure and work environment include verification, validation, monitoring, measurement, inspection, handling, storage, and distribution. Traceability activities are established to be commensurate with the products and processes.
Customer-Related Processes
These are the regulatory requirements covering the customers’ needs. In this case, ”customers” refers to the obvious patient/end-user of the device and includes anyone who comes in contact with the device. This may cover regulatory agencies, distributors, purchasing organizations, and shipping and logistics resources.
Design and Development Processes
Covered under this heading are all processes for the design and development of new devices, including: planning, inputs, outputs, reviews, verification, validation, transfer, change control, and files.
Purchasing
This step includes all purchasing processes ensuring that the purchased materials, components, and other products and services meet defined specifications. It includes defining criteria for supplier evaluation, ongoing performance, selection, and monitoring.
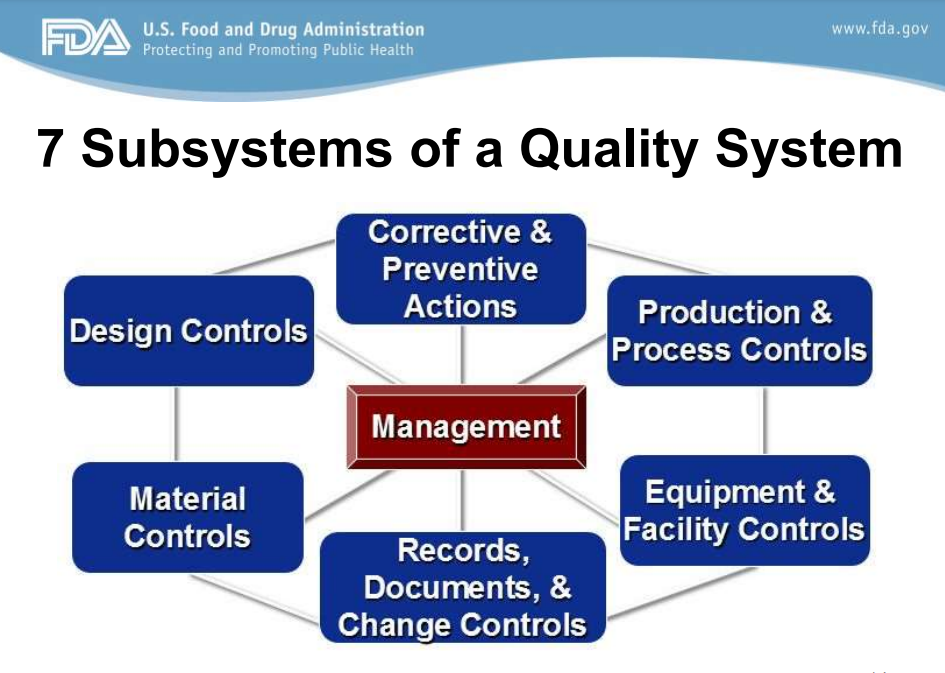
QMS According to FDA 21 CFR Part 820
Management Controls (MC)
The purpose of management controls is ultimately to provide adequate resources, ensure the establishment and effective functioning of the quality system, monitor the quality system, and make necessary adjustments.
To accomplish this purpose, the MC is required to establish a quality policy, plan, procedures, objectives, and organizational structure. It also must establish appropriate responsibility and authority, and appoint a management representative. In addition, the MC conducts management reviews and quality audits. It must have sufficient personnel with the necessary education, background, training, and experience.
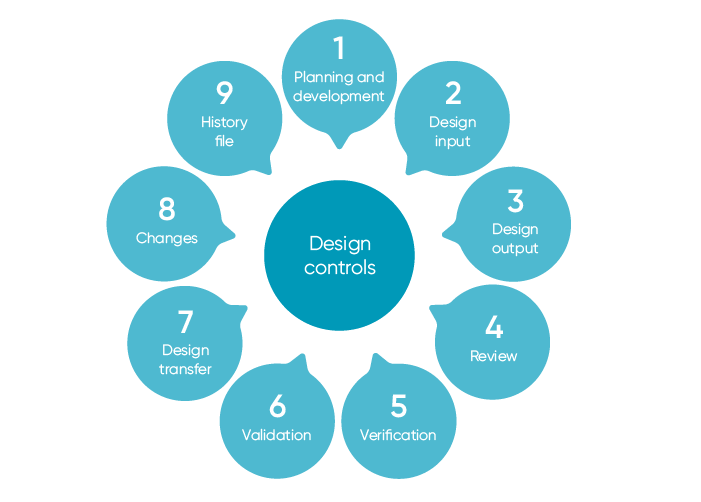
Design Controls
Design controls’ purpose is ultimately to control the entire design process and assure the device design meets user needs, intended uses, and specified requirements. To accomplish this, it must establish a plan to:
- Describe or reference design and development activities
- Identify design inputs
- Develop design outputs
- Verify that design outputs meet design inputs
- Validate the design (including software validation and risk analysis)
- Control design changes
- Review design results
- Transfer the design to production
- Compile a design history file
Production and Process Controls
The purpose of production and process controls is to manufacture products that meet specifications, develop processes that are adequate, validate (or fully verify the results) those processes, and monitor and control the entire manufacturing process.
Production processes must be monitored to ensure that the device conforms to its specifications. Statistical techniques that assess the acceptability of processes need to be identified. A valid statistical rationale must be employed to base sampling plans upon, while ensuring adequate sampling methods.
Corrective and Preventive Action (CAPA)
The purpose of CAPA is to standardize the response when corrective action needs to be taken (because something went wrong.) This might mean collecting information/data, identifying and investigating product and quality problems, and effecting corrective and preventive action. It also encompasses verifying or validating corrective and preventive actions, communicating corrective and preventive actions to appropriate personnel, and providing information for management review. All these activities must be documented.
Ultimately, employing a robust eQMS designed for the life sciences industry will save medical device manufacturers the headaches of meeting the standards and regulations to distribute devices in both the EU and the US.
eQMS is the Only Step You Need
Safety and quality regulations are becoming more complex and compliance standards more stringent. Many companies are facing difficulties in keeping up with tougher regulations due to a lack of adequate resources and having few internal initiatives to stay current with regulatory changes. Maintaining correct compliance can be as costly as it is complicated. It’s estimated that USD 4.5 billion was spent on the global medical device regulatory market in 2020, with expected expansion at a CAGR of 8.6% from 2021 to 2028.
Implementing regulatory standards requires a combination of people, processes, and technology. Since these standards are vital to every aspect of medical device manufacturer, it is essential to deploy a comprehensive, professional solution. Thankfully for growing and established companies alike, the advent of digital QMS solutions has eased the burden of QMS regulatory compliance.
Implementing a QMS is an essential requirement for any medical device company. With the complexity involved and the rapidly changing regulatory requirements, many medical device manufacturers are choosing to use digital quality management systems. Leading the field in eQMS systems is Dot Compliance. Dot Compliance’s cloud-based platform, built upon Salesforce, allows companies to scale their operations while maintaining full compliance seamlessly.
Also, by working with an external provider that specializes in eQMS, manufacturers know that they have expert input and immediate response to any regulatory changes without them needing to take any independent steps. With Dot Compliance, navigating regulatory compliance becomes easy for medical device manufacturers and life science companies.
